
Bergithe Eikeland Oftedal og Eystein S. Husebye.
Bergithe Eikeland Oftedal, PhD, Universitet i Bergen.
Eystein S. Husebye, professor, overlege, Haukeland universitetssykehus og Universitetet i Bergen
Organspesifikke autoimmune sykdommer er svært vanlige og en viktig årsak til redusert livskvalitet og død. De rammer ofte i ung alder og kan ha sterk familiær opphopning. En og samme person kan affiseres av flere sykdommer, noe som kalles et autoimmunt polyendokrint eller polyglandulært syndrom (APS). Tradisjonelt har en antatt at risiko for utvikling av disse sykdommene er knyttet til varianter i en rekke immungener i interaksjon med miljøfaktorer. Nye funn ty der på at monogene former forekommer hyppigere enn vi tidligere har antatt. Nå kan ny diagnostikk hjelpe oss å stille diagnosen.
Organ spesifikke autoimmune sykdommer rammer ofte endokrine organer og innbefatter sykdommer som autoimmune tyreoideasykdommer både med hypotyreose og hypertyreose som resultat (blant annet Graves sykdom), type 1 diabetes, binyrebarksvikt, ovariesvikt, hypofysesvikt og hypoparatyreoidisme i fallende frekvens. Assosiert er også sykdommer i en rekke ikke-endokrine organer som hud (vitiligo), hår (alopesi), og gastrointestinaltraktus (autoimmun gastritt og cøliaki) [1]. Som gruppe rammer disse sykdommene i ung alder, kvinner oftere enn menn, og de fører ofte til redusert livskvalitet og økt dødelighet. Et autoimmunt polyendokrint syndrom (APS) foreligger når en person har flere av disse sykdommene.
Ved APS type-3 (APS-3) har pasienten autoimmun thyreoideasykdom kombinert med type 1 diabetes eller andre organ-spesifikke sykdommer som autoimmun gastritt, vitiligo eller alopesi. Ved APS type 2 (APS-2) er den definerende komponenten autoimmun binyrebarksvikt (Addisons sykdom) kombinert med autoimmun thyreoideasykdom og/eller type-1 diabetes. APS-2 og APS-3 er assosiert med de samme immungenene, hvor først og fremst HLA («human leukocyte antigen») varianter i interaksjon med eksterne miljøfaktorer ligger bak. Hvorfor en person får type 1 diabetes og en annen Addisons sykdom og en tredje begge, kan bero på ulike miljøfaktorer eller rett og slett tilfeldigheter. Det har derfor vært foreslått å samle APS-2 og -3 til en gruppe og kalle dem APS-2 [2].
APS type 1 (APS-1) skiller seg fra de øvrige formene ved å være monogent arvelig og svært sjelden (1:80 000). APS-1 er et av noen få eksempler på at et enkelt gen kan gi et auto-immunt polyglandulært syndrom [3]. Studier av APS-1 og andre monogene sykdommer i immunsystemet har vært ekstremt nyttig, ikke bare for å forstå sykdomsmekanismene, men også for å forstå normal fysiologi og for å utvikle ny terapi [4].
Klinisk defineres APS-1 med funn av to av de tre hovedkomponentene hypoparatyreoidisme, primær binyrebarksvikt (Addisons sykdom) og kroniske ikke-invasive kandidainfeksjoner på slimhinner, hud og negler. En av de tre hovedkomponentene er tilstrekkelig for diagnose dersom et søsken allerede er diagnostisert, eller dersom mutasjoner er funnet på begge allelene i AIRE [5] (beskrives nærmere lenger ned i teksten). APS-1 bør mistenkes hvis en av de tre hovedkomponentene presenteres før pasienten er 20 år eller om det foreligger uvanlige organ-spesifikke manifestasjoner som emaljehypoplasi, keratitt eller ovariesvikt i ung alder [6]. Majoriteten av kvinnelige APS-1 pasienter går i menopause før 30 års alder. En rekke gastrointestinale komponenter er også vanlig. Rundt en tredjedel rammes av autoimmun enteritt med malabsorpsjon, kronisk diare, av og til avløst av perioder med hardnakket obstipasjon. Autoimmun hepatitt rammer i ung alder og kan være dødelig. Autoimmun gastritt med pernisiøs anemi er også vanlig. I tillegg utvikler mange såkalte ektodermale komponenter. Emaljehypoplasi av blivende tenner ses hos om lag 80 prosent av pasientene [6]. Mange utvikler vitiligo og alopesi. Keratitt kan lede til blindhet og sialoadenitter til tørre øyne og nedsatt spyttsekresjon (Figur 1).
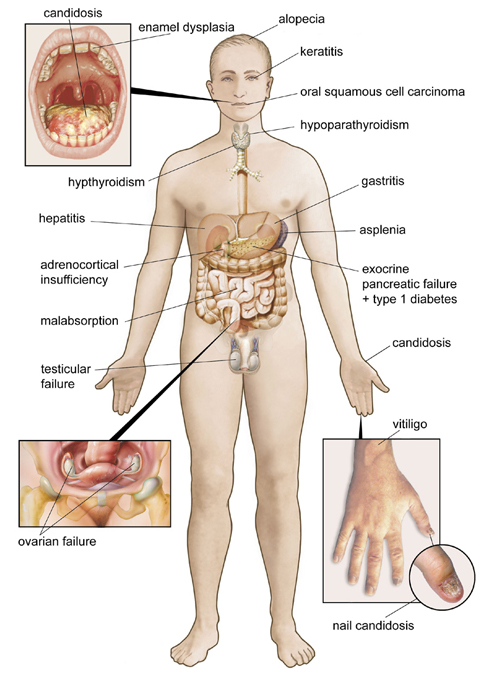
Figur 1. Illustrasjonen av de ulike sykdomskomponentene ved APS-1. Gjengitt fra ref 5, med tillatelse.
I Norge er prevalensen rapportert til 1:80 000 [7], et tall som regnes som representativt for de fleste andre land og som ganske sikkert er en underestimering. I noen befolkninger er betydelig høyere prevalenssifre rapportert som i Finland (1:25000), på Sardinia (1:14000) og blant Persiske jøder (1:9000). Diagnosen vanskeliggjøres ved stor variasjon i sykdomsbildet, også innen samme familie. Sykdommen presenterer seg typisk tidlig i barnealder, men nye autoimmune manifestasjoner kan tilkomme gjennom hele livet [6]. Det kan gå lang tid mellom utviklingen av nye komponenter og sykdommen kan også starte i ungdomsår eller tidlig voksen alder. Det kan derfor i starten være vanskelig å vite om pasienten har den «vanlige» formen for organ-spesifikk autoimmun sykdom eller om APS-1 foreligger.
Kroniske soppinfeksjoner er ofte det første tegnet på APS-1 og opptrer typisk i det første leveåret med infeksjoner med Candida Albicans på negler, hud og i slimhinner i munnen, spiserøret og vagina. Hos finske pasienter ble en av seks diagnostisert med kronisk soppinfeksjon før ett års alder, og dette tallet økte til 98 % ved 30 års alder [8]. Vi finner liknende sifre blant de norske pasientene [6]. Infeksjonene varierer i alvorlighetsgrad, men kommer ofte tilbake og kan være vanskelig å behandle. Vedvarende infeksjoner disponerer for plateepitelkarsinom i munnhule og stenoser i spiserøret. Det er derfor viktig med god soppbehandling og å unngå røyking [5].
Hypoparatyreoidisme er den nest vanligste manifestasjonen i APS-1 og opptrer hos 85 % av pasientene ved fylte 30 år [8]. Ved ikke-medfødt hypoparatyreoidisme skal APS-1-diagnosen aktivt utelukkes (se nedenfor). Dette er vanligvis den første endokrine manifestasjonen, og den opptrer noe hyppigere hos kvinner enn hos menn. Hypoparatyreoidisme skyldes autoimmun ødeleggelse av paratyroidiavevet, noe som resulterer i redusert plasma-kalsium med parestesier og kramper. Typiske laboratoriefunn er lav parathyreoideahormon, lav kalsium og høy fosfat uten nyresvikt.
Autoimmun Addisons sykdom opptrer gjerne etter kroniske soppinfeksjoner og hypoparatyreoidisme. Blant finske pasienter opptrer binyrebarksvikt i 78 % av tilfellene innen 30 års alder [8], i Norge hos 63 % uansett alder [6]. Addisons sykdom karakteriseres av trøtthet, salthunger, vekttap og økt pigmentering av hud og slimhinner. Diagnosen stilles ved å påvise lav serum kortisol i en morgenprøve kombinert med forhøyet ACTH eller manglende stigning i kortisol etter tilførsel av synachten (ACTH-analog). Substitusjonsbehandling med hydrokortison/kortisonacetat og fludrokortison vil i stor grad normalisere mortalitetsratene. Om Addisons sykdom opptrer i ung alder (under 20 år) skal en tenke på om APS-1 kan foreligge.
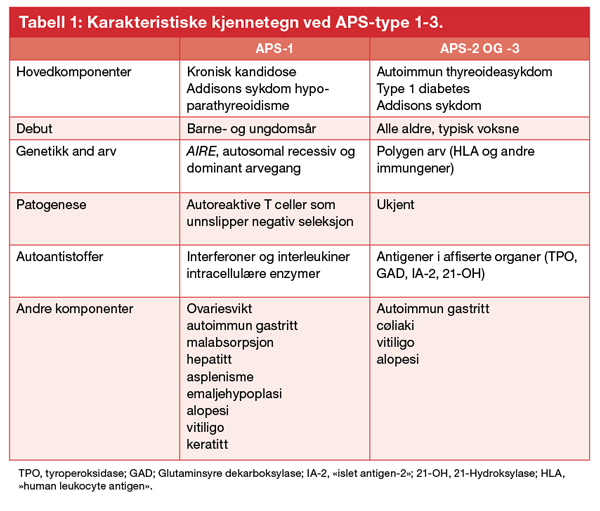
Siden pasienter med APS-1 ikke etablerer immuntoleranse mot selv-antigenene, vil B-celler og plasma-celler fra pasienter typisk produsere flere ulikeautoantistoffer mot intracellulære enzymer i affiserte organer og mot enkeltkomponenter i immunforsvaret, blant annet cytokiner og interlevkiner (Tabell 1). Man deler derfor gjerne autoantistoffene i organ-spesifikke og cytokin-autoantistoffer, men felles for begge gruppene er at de ofte opptrer før kliniske symptomer og kan brukes i diagnostikk av pasienter med APS-1. Noen av autoantistoffene er relativt spesifikke for APS-1, mens andre finnes ved de vanlige sykdomskomponentene som inngår i APS-2 og APS-3 [1]. De organ-spesifikke autoantistoffene gjenkjenner typisk intracellulære enzymer som er spesifikke for organet de utrykkes i. Autoantistoffer mot enzymet 21-hydroksylase (21-OH) som uttrykkes i binyrebarken, er en spesifikk markør for autoimmun Addisons sykdom, mens autoantistoffer mot «side-chain cleavage» enzymet (SCC) er markør for både Addisons sykdom og ovariesvikt; dette enzymet uttrykkes i både binyrebarken og ovarier. Autoantistoffer mot 21-OH er tilstede fra måneder til år før andre biokjemiske og kliniske tegn på binyrebarksvikten inntrer [9]. I og med at binyrebarksvikt er så hyppig ved APS-1 er det klinisk indisert å følge autoantistoffer hos pasientene og regelmessig teste binyrefunksjonen når autoantistoffer foreligger. Tilsvarende er autoanti-stoffer mot NACHT leucine-rich-repeat protein 5 (NALP5), et protein med ukjent funksjon og en markør som for hypoparatyreoidisme, uttrykt nesten utelukkende i paratyreoideakjertlene [10].
Autoantistoffer mot aromatisk L-aminosyre dekarboksylase (AADC) er vanlig ved autoimmun hepatitt og vitiligo [11], mens tryptofanhydroksylase, et enzym i serotoninbiosyntesen som det finnes mye av i tarm, er et autoantigen assosiert med autoimmun enteropati [12].
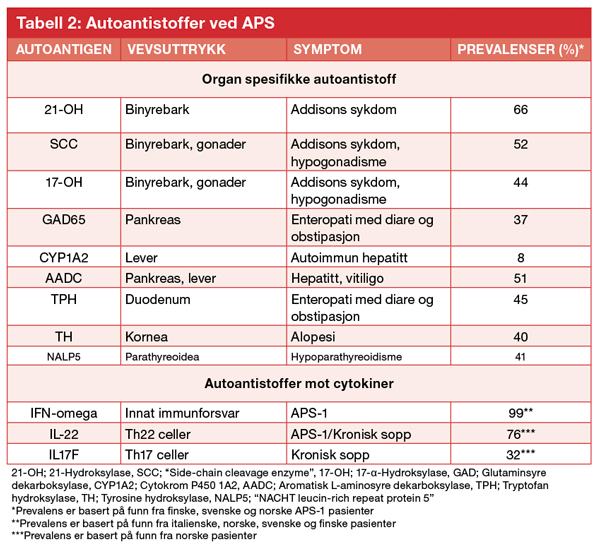
Autoantistoffer mot cytokiner og interleukiner er mer spesifikke for APS-1, og spesielt anti-interferon omega antistoffer detekteres hos nesten alle pasientene (> 99 %) (Figur 2) [13]. Denne analysen kan derfor brukes som en screeningmarkør for diagnosen APS-1 som siden kan bekreftes ved sekvensering av autoimmune regulator (AIRE). Autoantistoffer mot interleukin 17 og 22 er beskrevet, med en mulig link til de kroniske soppinfeksjonene man ser hos disse pasientene [14]. Også anti-interferon omega opptrer før kliniske symptomer, og er funnet så tidlig som ved 7 måneders alder. Opphavet og rollen til disse autoantistoffene er fortsatt uklar, og er også beskrevet hos pasienter med myasthenia gravis med tymom og milde former for immundefekter på bakgrunn av mutasjoner i rekombinasjonsgenene RAG1 og RAG2. Felles for disse tilstandene er en defekt funksjon/uttrykk av AIRE i thymus og thymomer.
AIRE
AIRE-genet ble identifisert som den underliggende årsaken til APS-1 i 1997. Genet ligger på kromosom 21 og har 14 eksoner som koder for et 545 aminosyre langt protein, som er aktivt som en dimer eller multimer. Det er per i dag rapportert 115 mutasjoner i AIRE, og mutasjoner er funnet i 95 %
av de kliniske tilfellene [15]. AIRE utrykkes i thymus, i celler kalt thymiske medullære epiteliale celler. AIRE spiller en viktig rolle i negativ seleksjon av selv-reaktive T-celler og utviklingen av T-regulatoriske celler. AIRE er ikke en tradisjonell transkripsjonsfaktor, men en transkripsjonsregulator som stimulerer uttrykket av organ-spesifikke proteiner som insulin og amelogenin, proteiner som vanligvis ikke uttrykkes i thymus. Slik er AIRE essensiell for å eksponere T-celler under modning for peptider fra et stort repertoar av kroppens proteiner slik at autoreaktive T-celler kan fjernes fra det immunologiske repertoaret. Fungerer ikke AIRE, vil autoreaktive T-celler slippe ut i periferien der de senere kan bidra til autoimmun sykdom [1].
En rekke studier har undersøkt om varianter og mutasjoner i AIRE er assosiert med vanlige autoimmune sykdommer som type 1 diabetes, autoimmun thyreoideasykdom, alopesi og reumatoid artritt uten å komme fram til klare sammenhenger [16]. De fleste studiene har vært små og basert på «single nucleotid polymorphism» (SNP) analyser, ikke på sekvensering av genet. Nylig har vi beskrevet familier med et mildere APS-1-liknende sykdomsbilde assosiert med bare en mutasjon i AIRE, som alle grupperer seg til et avgrenset område av genet, det såkalte PHD1- (plant homeo-domain 1) [17]. Mutasjoner her ser ut til å ha en dominant negativ effekt på det friske allelet og dermed inhibere AIRE sin funksjon. Familier med slike mutasjoner har en klinisk fenotype som varierer fra klassisk APS-1 til isolert organ-spesifikk autoimmunitet, med overvekt av vitiligo og pernisiøs anemi. Når en mutasjon er tilstrekkelig til å utvikle sykdom er arvegangen dominant med nedarving fra en generasjon til en annen. Tilgjengelige databaser viser at de dominante mutasjonene i PHD1 har en frekvens på minst en av tusen [17]. Det kan derfor se ut som at familiær opphopning av autoimmunitet i noen tilfeller skyldes mutasjoner i AIRE.
I 1996 ble det norske registeret for organspesifikke autoimmune sykdommer (ROAS) etablert ved Haukeland universitetssykehus og Universitetet i Bergen (www.haukeland.no/roas). Registeret som er et nasjonalt medisinsk kvalitetsregister samler informasjon fra pasienter med endokrine autoimmune sykdommer, spesielt pasienter med APS-1, Addisons sykdom og hypoparatyreoidisme. ROAS inneholder informasjon vedrørende sykdomskomponenter, pågående behandling, autoantistoffprofiler og genvarianter for flere gener relevant for autoimmune sykdommer. Biobanken inkluderer blod, sera, DNA, mononukleære celler fra blod, tårer og spytt, regelmessig samlet inn fra pasientene. Både karakteriseringen og samlingen av biologisk materiale er et pågående arbeid som inkluderer et nettverk av endokrinologer ved sykehus i alle helseregioner i Norge.
APS-1 er en uvanlig, men under-diagnostisert sykdom, karakterisert av organ-spesifikk autoimmunitet mot en lang rekke organer. Diagnosen stilles klinisk ved påvisning av to av de tre hovedkomponentene binyrebarksvikt, hypoparatyreoidisme og kroniske soppinfeksjoner eller påvisning av mutasjoner i AIRE og en sykdomskomponent. Autoantistoffer mot interferon omega er en god screeningmarkør ved klinisk mistanke. Dominant ikke-klassisk APS-1 kan forklare tilfeller av familiær opphopning og er karakterisert ved dominant arvegang og høy frekvens av vitiligo og pernisiøs anemi. Det er viktig å informere om sykdomsbildet til leger og tannleger med ulike spesialiteter slik at pasientene kan diagnostiseres tidlig før livstruende komplikasjoner oppstår.
Forfatterne har ingen interesse-konflikter å rapportere



