
Mathis Korseberg Stokke.
Av Mathis Korseberg Stokke, Klinikk for medisin, Lovisenberg Diakonale Sykehus; Institutt for eksperimentell medisinsk forskning, Oslo universitetssykehus,
Ullevål og Universitetet i Oslo; KG Jebsen, Cardiac Research Center og Center for Heart Failure Research, Universitetet i Oslo. e-post: m.k.stokke@medisin.uio.no
Artikkelen er tidligere publisert i Hjerteforum nr 3, 2015 og trykkes med tillatelse.
Ventrikulære ekstrasystoler forekommer hyppig i en klinisk hverdag. Denne artikkelen tar for seg årsaksforhold og mekanismer. Dokumentasjonen for at ventrikulære ekstrasystoler har patologisk betydning er begrenset. Artikkelen drøfter dette og aktuell behandling.
Summary in English
Premature Ventricular Complexes (PVCs) are a highly prevalent phenomenon in everyday clinical practice. Nevertheless, the observation of PVCs is often met with uncertainty with regard to clinical significance, work-up, and indications as well as alternatives with regard to treatment. This clinical uncertainty is contrasted by the increasing insight in the fundamental mechanisms underlying PVCs. The task for the clinician is to separate the situations in which PVCs occurs as a benign phenomenon in an otherwise healthy individual, from the situations in which PVCs could indicate underlying cardiac disease, or even act a cause or trigger of such disease. This review introduces the reader to some of these insights from basic research, as well as clinical data, and the lack thereof that can guide clinical decision making when confronted with PVCs.
A full version of this article with extended references is available on request.
Ventrikulære ekstrasystoler (VES) er et fenomen alle klinikere må ta stilling til, enten som et tilfeldig bifunn, som hovedårsak til pasientens plager, som prognostisk faktor, som uttrykk for ugunstig påvirkning av hjertet eller som tegn på manifest hjertesykdom. Utfordringen er å plassere pasientens VES i den rette av disse kategoriene og avgjøre om videre utredning og/eller behandling er nødvendig. I mange situasjoner har imidlertid VES usikker betydning, og på grunn av manglende retningslinjer varierer praksis for utredning og behandling. Denne artikkelen gir en kort innføring i det patofysiologiske og empiriske grunnlaget for å forstå, utrede og behandle VES i forskjellige sammenhenger og pasientgrupper.
VES er prematur aktivering av myokard fra et utgangspunkt i ventriklene. I EKG ses dette som QRS-komplekser uten forutgående P-bølge og med en annen konfigurasjon enn ved aktivering fra et supraventrikulært fokus, f.eks. sinusrytme (figur 1). QRS-konfigurasjonen avhenger av utgangspunktet for aktiveringen og ledningen gjennom myokard og er oftest breddeforøket. I motsetning til erstatningsslag kommer QRS-komplekset ved VES prematurt i forhold til et normalt sinusslag og følges ofte av en kompensatorisk pause. Grunnen til pausen er at VES ikke depolariserer sinusknuten, som derfor fortsetter med sin egen aktiveringsfrekvens. Dermed blir RR-intervallet mellom det siste slaget før VES og det første slaget etter, dobbelt av RR-intervallet ved den foregående sinusrytmen. Pasientene kjenner det første normale slaget etter VES best siden pausen medfører bedre fylling og dermed «post-systolisk potensiering». Denne effekten er kraftigst i friske hjerter.
VES kan oppstå fra alle de tre prinsipielle mekanismene for takyarytmier: automati, trigget aktivitet og reentry-fenomener. En grunnleggende forståelse av disse mekanismene er til hjelp for forståelsen av VES i forskjellige kliniske tilstander og situasjoner. For innføring i basal og klinisk elektrofysiologi og arytmologi anbefales bøkene til hhv. Donald M. Bers (Excitation-Contraction Coupling and Cardiac Contractile Force, 2001) og Antoni Bayes de Luna (Clinical arrhythmology, 2011).
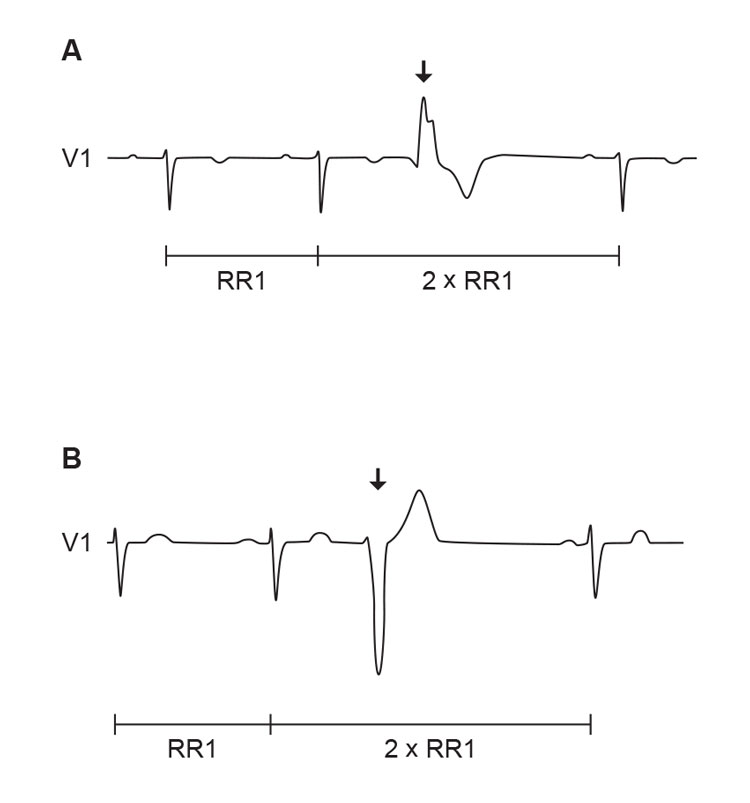
Figur 1. VES gjenkjennes i EKG som et prematurt QRS-kompleks uten forutgående P-bølge som er breddeforøket og har annen konfigurasjon enn supraventrikulære slag. VES etterfølges oftest av en pause. A) VES fra fokus i venstre ventrikkel, B) VES fra fokus i høyre ventrikkel (Ill. ved A. K. Stokke 2015).
Automati er mekanismen som gir sinusknuten dens pacemakeregenskaper. Også annet vev i hjertet har latente pacemakeregenskaper, men disse «latente pacemakerne» undertrykkes av aktivering fra sinusknuten. Mekanismene for automati er sannsynligvis de samme for alle vevstyper med latente pacemakeregenskaper, men med forskjellig aktiveringsfrekvens i forskjellige celler. Celler i andre deler av hjertet enn sinusknuten kan overta aktiveringen av hjertet hvis de får en høyere aktiveringsfrekvens. Dette kalles abnorm eller unormal automati. Figur 2 beskriver hvordan normal ionehomeostase i kontraktile hjertemuskelceller skiller seg fra de to mekanismene som kan bidra til automati i nodale celler og ved patologiske tilstander også i annet vev.
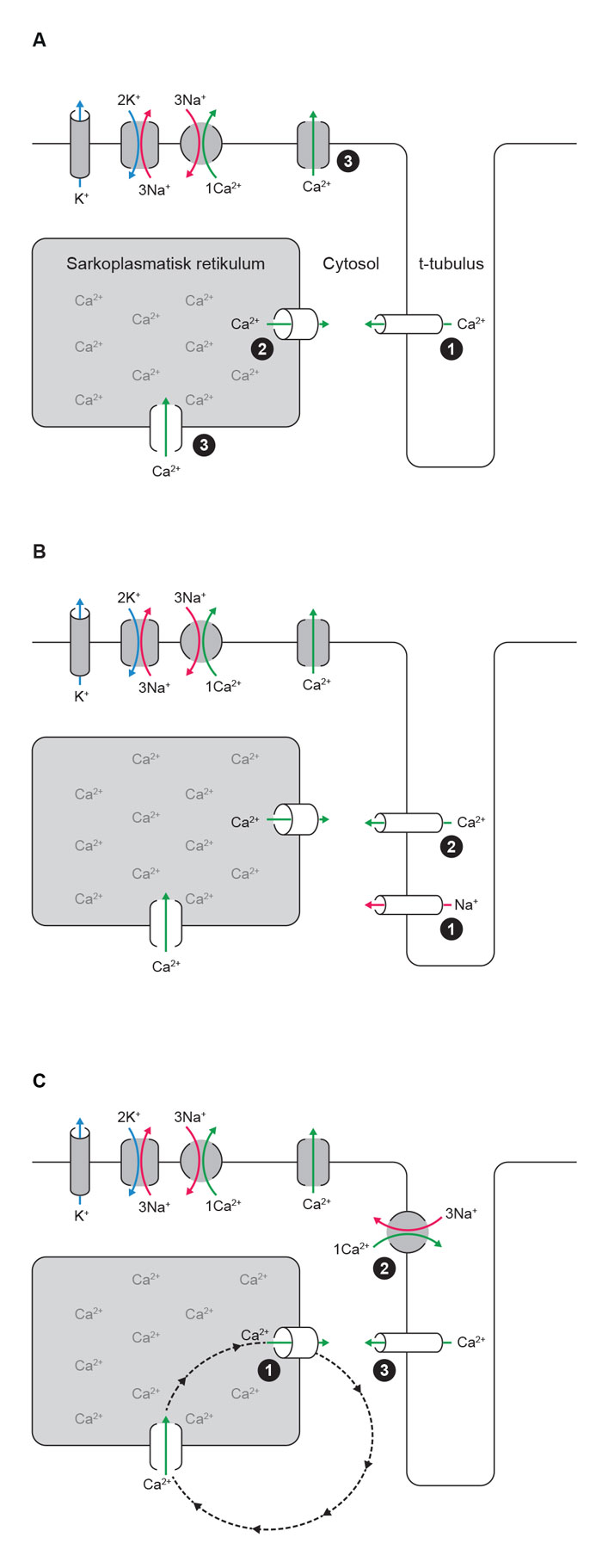
Figur 2. A) Hovedkomponenter i normal ionesyklus i ventrikkelmuskelceller. 1. Ved depolarisering av cellemembranen åpner L-type Ca2+-kanal og slipper en liten mengde Ca2+ inn i cytosol. 2. Ca2+ binder til Ca2+-frislippskanalen RyR2, som dermed åpner og slipper en større mengde Ca2+ ut i cytosol. Ca2+ binder til myofilamentene og leder til kontraksjon. 3. For relaksasjon tas Ca2+ tilbake opp i det sarkoplasmatiske retikkelet og pumpes ut av cellen. B) Automati ved ivabradin-følsom «funny current», også kalt «pacemakerstrøm». 1. Pacemakerstrømmen leder kationer inn i cellen og leder til langsom depolarisering av cellemembranen inntil 2. terskelverdien for aksjonspotensialet nås, f.eks. ved aktivering av L-type Ca2+ kanal. C) Automati ved «Ca2+-klokke». 1. Ca2+ lekker fra det sarkoplasmatiske retikkelet, og 2. utveksles med Na+. Dette medfører en netto innstrømming av positive ladninger i cellen og kan til slutt 3. depolarisere cellen tilstrekkelig til aktivering av f.eks. L-type Ca2+-kanal. Frekvensen av slik aktivering vil avhenge av frigjøring og reopptak av Ca2+ i det sarkoplasmatiske retikkelet (stiplet sirkel) (Ill.ved A. K. Stokke 2015).
Proteinene som er involvert i ionesyklus ved automati kan påvirkes av sympatisk aktivitet. Dermed kan aktiviteten i det autonome nervesystemet regulere aktiveringsfrekvensen i pacemakercellene. De samme proteinene er imidlertid også følsomme for hypoksi og iskemi. Slik kan celler i iskemisk vev få en aktiveringsfrekvens som er høyere enn sinusknutens, og føre til prematur aktivering, altså VES. Dette antas å være en av mekanismene for VES og idioventrikulær rytme i den tidlige fasen av akutt koronarsyndrom og etter reperfusjon. I ventriklene er det spesielt purkinjefibrene som kan utvikle slik aktivitet.
Trigget aktivitet skyldes ikke pacemakeregenskaper, men avhenger av den forutgående aktiveringen av cellene. Relasjonen til den forutgående aktiveringen er imidlertid kompleks, og forskjellige mekanismer kan antagelig spille en rolle ved trigget aktivitet utløst av hhv. takykardi og bradykardi (1). I motsetning til den gradvise depolariseringen som er grunnlaget for automati, ses trigget aktivitet som en relativt rask depolarisering i repolariseringsfasen (fase 2 og 3) eller hvilefasen (fase 4) av aksjonspotensialet (figur 3A). Depolarisering i repolariseringsfasen kalles tidlige etterdepolariseringer og ses spesielt i situasjoner med forlenget aksjonspotensial og dermed lang QT-tid (figur 3B). Dette kan f.eks. illustreres av Timothy-syndrom som skyldes mutasjoner i L-type Ca2+-kanalen og medfører både lang QT-tid, tidlige etterdepolariseringer og ventrikulære arytmier.
Mekanismen for depolarisering i hvilefasen av aksjonspotensialet (sene etterdepolariseringer, figur 3B) avhenger av andre proteinene. Normalt skal Ca2+ strømme ut av det sarkoplasmatiske retikkelet når cellen aktiveres av sinusstyrte impulser (figur 2A). Ca2+-frislippskanalen i retikkelet, RyR2, kan imidlertid bli dysfunksjonell pga. iskemi, sympatisk aktivitet eller mutasjoner. Slike faktorer kan endre aktiviteten til RyR2 og føre til at Ca2+ strømmer ut også i fase 4 av aksjonspotensialet. Slik unormal Ca2+-frigjøring kan resultere i VES ved iskemi og reperfusjon, men illustreres tydeligst ved katekolaminerg polymorf ventrikkeltakykardi (CPVT). RyR2-mutasjonene i denne sykdommen medfører lekkasje av Ca2+ i fase 4, etterdepolariseringer og hyppige VES under fysisk aktivitet.
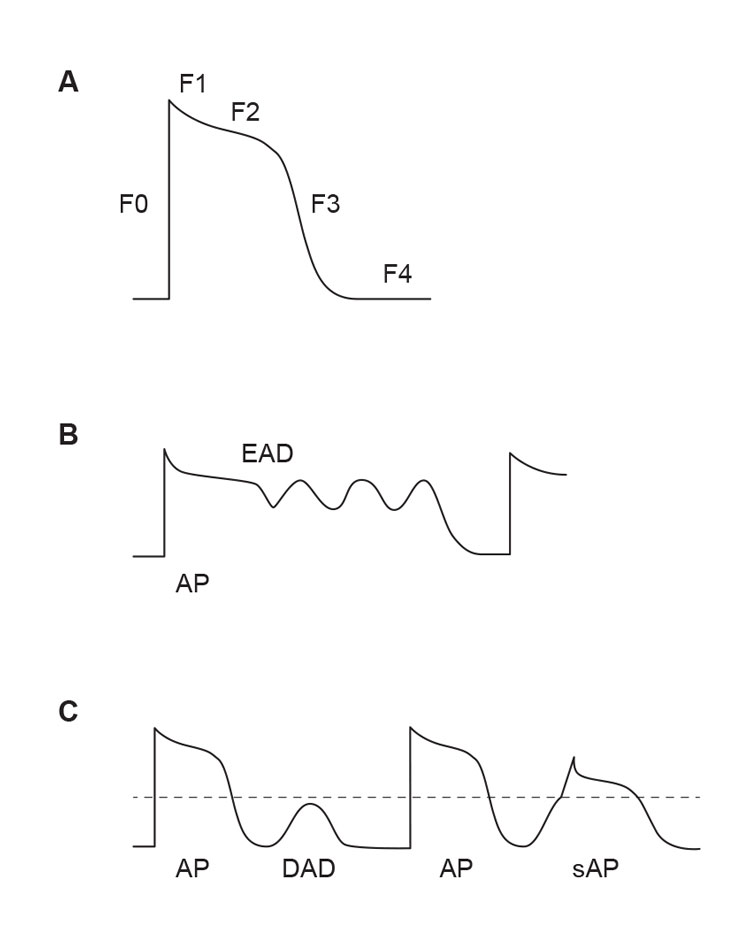
Figur 3. Trigget aktivitet. A) Fasene i ventrikkelcellenes aksjonspotensial. B) Tidlige etterdepolariseringer (early afterdepolarisations, EADs) oppstår typisk ved forlenget aksjonspotensial i fase 2 eller 3. C) Sene etterdepolariseringer (delayed afterdepolarisations, DADs) oppstår i fase 4 og kan utløse aksjonspotensialer hvis de når terskelverdien (stiplet linje). (Ill. ved A. K. Stokke 2015).
Både automati og trigget aktivitet er fenomener på cellenivå. Den relativt svake strømmen som oppstår fra enkeltceller vil ikke kunne aktivere hele myokard slik man ser ved VES. Forsøk med data fra kaninhjerter anvendt i matematiske modeller viser at i størrelsesorden 800 000 celler må depolarisere synkront for å aktivere myokard (2). Når man endret modellen slik at den etterliknet myokard ved uttalt hjertesvikt ble dette imidlertid redusert til ca. 4000 celler. Sympatisk stimulering synkroniserer også prosessene som fører til sene etterdepolariseringer i flere celler. Dermed øker sannsynligheten for at trigget aktivitet inntreffer samtidig i et tilstrekkelig antall celler til å gi VES.
Reentry-fenomener skyldes at forskjellige områder av myokard leder impulser med forskjellig hastighet eller med forskjellig refraktærperiode. Dermed oppstår muligheten for at impulser som på et gitt tidspunkt ledes i ett område av myokard, danner en sluttet krets med et annet område som kan aktiveres på et senere tidspunkt (figur 4). Dette er antagelig den vanligste mekanismen for arytmi etter etablering av arrvev som følge av infarkt og kan involvere ledningssystemet eller deler av myokard. Arrvev eller strukturelle endringer av myokard medfører permanent endret ledningshastighet og således et statisk substrat for reentrykretser. Substratet kan imidlertid også være dynamisk som følge av variasjoner i ionekanalaktivitet, elektrolyttforstyrrelser, iskemi m.m.
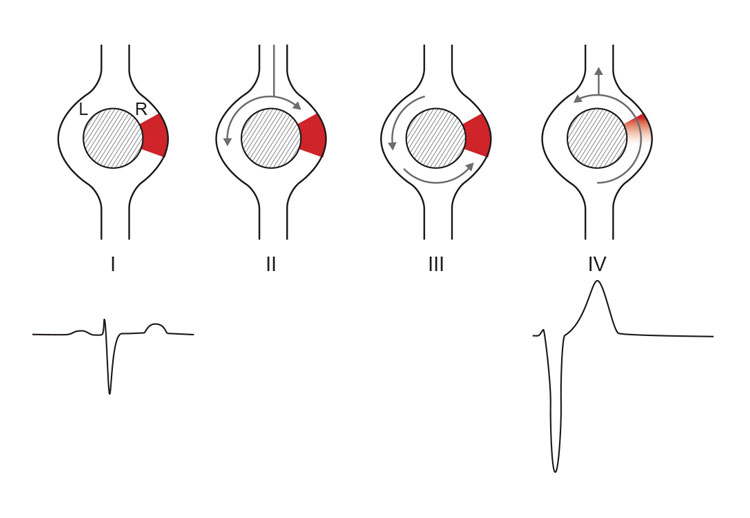
Figur 4. Reentrykretser betinget av forskjellig ledningshastighet. Område «R» i figuren har langsommere ledning enn område «L». Dermed er R refraktær lengre enn L. Når refraktærperioden er overstått, kan man imidlertid risikere at impulser som har passert gjennom område L, kan passere retrograd gjennom og dermed initiere en reentrykrets. (Ill. ved A. K. Stokke 2015).
VES kan initiere vedvarende ventrikkeltakykardier. Bayes de Luna hevder t.o.m. at «de fleste» ventrikkeltakykardier startes av en VES. Utfordringen ligger imidlertid i å forutsi hvor sannsynlig dette er for den enkelte pasienten. Risikoen for at VES trigger en vedvarende arytmi avhenger av statiske og dynamiske substrater for reentrykretser. Den høye risikoen for ventrikkeltakykardi og plutselig død ved alvorlig hjertesvikt illustrerer dette: Hjertesvikt medfører endringer i ionekanalekspresjon og -aktivitet, endret cellulær og ekstracellulær ionehomeostase samt regional iskemi og fibrose. Dermed øker sannsynlighet for både automatisme og trigget aktivitet samtidig som både statiske og dynamiske substrater for reentrykretser er til stede.
De senere årene har man blitt oppmerksom på at VES også kan forårsake svekket kontraktil myokardfunksjon. Sannsynligvis er dette kun relevant ved en vedvarende svært høy frekvens av VES, dvs. 10 000 – 20 0000 VES per døgn (3). Den kausale sammenhengen mellom en såpass høy frekvens av VES og svekket kontraktil funksjon ble overbevisende dokumentert i et forsøk med hunder utsatt for pacemakerstimulerte VES i bigemini (4). Etter 12 uker med slik stimulering var venstre ventrikkels ejeksjonsfraksjon redusert med 45 %. Dette var imidlertid fullt reversibelt etter 4 uker når pacemakeren ble slått av og hundene returnerte til sinusrytme. Tilsvarende funn er gjort hos pasienter etter vellykket ablasjon for svært hyppige VES (5). Mekanismen for slik «VES-indusert kardiomyopati» er ikke klarlagt, men involverer sannsynligvis uhensiktsmessig hemodynamikk, lokalt strekk og dyssynkroni i myokard, endret ekspresjon og aktivitet av ionekanaler og metabolske forandringer. Hvorvidt dette fenomenet er det samme som takykardi-indusert kardiomyopati, er omdiskutert.
Som isolert fenomen i EKG kan VES være en tilfeldig observasjon uten symptomer, en forklaring på palpitasjoner uten prognostisk betydning, en markør for hjertesykdom, en årsak til kardiomyopati eller en potensiell trigger for maligne arytmier. Kliniske data gir oss til en viss grad støtte for å identifisere gruppen hvor VES kun ses som et tilfeldig funn eller gir palpitasjoner uten prognostisk betydning. Ved mistanke om VES som uttrykk for annen hjertesykdom, bør dette avklares på vanlig måte, mens risikoen for VES-indusert kardiomyopati kan vurderes ut fra frekvensen av VES.
Begrepet «hyppige VES» brukes ofte og kan være inngangsporten til utredning eller behandling. Dette begrepet indikerer imidlertid en kvantitativ tilnærming som har dårlig dekning i litteraturen. For friske unge siteres ofte mer enn 50 VES per døgn som hyppig basert på Holteropptak fra en liten gruppe amerikanske studenter på 70-tallet (6). Senere studier har vist at hyppigheten av VES varierer med kjønn, alder og sykdomsstatus (7). VES er et vanlig fenomen også hos «hjertefriske», og normalvariasjonen for antall VES per døgn er sannsynligvis stor. Selv i en gruppe pasienter med normal arbeidsbelastning, ekkokardiografi, høyre- og venstrekateterisering samt koronarangiografi hadde 39 av 101 mer enn 1 VES ved 24-timers Holter-registrering, mens 4 av 101 hadde mer enn 100 VES (7).
Et annet problem har vært å avklare hvorvidt VES i seg selv kan brukes som en selvstendig risikofaktor for sykdom og død. En metodemessig meget godt utført meta-analyse publisert i 2013, fant at i den «generelle befolkningen» uten kjent hjertesykdom, er hyppige VES assosiert med økt risiko for plutselig hjertedød (8). En svakhet ved denne studien er imidlertid for det første at de 11 studiene som ble inkludert i analysen, definerte hyppige VES forskjellig, og for det andre at ingen av studiene ekskluderte pasienter med hjertesykdom på annen måte enn ved anamnese. En annen meta-analyse konkluderer på samme måte at hyppige VES nok er en risikofaktor for kardiovaskulær død, men om dette egentlig skyldes underliggende hjertesykdom vites ikke (3). Én studie som kun inkluderte pasienter med normal ekkokardiografi og arbeidsbelastning, fant ingen assosiasjon mellom VES og død uansett årsak etter gjennomsnittlig 6,5 års oppfølging (9).
I forhold til den noe uavklarte betydningen av VES som isolert fenomen i hvile eller under Holter-monitorering hos personer uten kjent hjertesykdom, er det bedre dokumentert at VES som fremprovoseres under eller etter belastning, har prognostisk betydning. Jouven et al. inkluderte menn mellom 42 og 53 år uten kjent hjertesykdom basert på anamnese og EKG (10). Forekomst av to eller flere VES i serie eller mer enn 10 % VES i løpet av en 30 sekunders periode under AKG eller i recoveryfasen ble definert som hyppige VES. I løpet av en oppfølgingsperiode på 23 år hadde menn med hyppige VES en 2,7 ganger høyere risiko for død av kardiovaskulær årsak sammenliknet med dem som ikke hadde slike VES. VES eller ventrikulære takykardier i recoveryfasen etter AKG som prediktor for død uansett årsak ble også gjenfunnet i en annen studie (11).
Flere grupper av pasienter med etablert hjertesykdom har økt hyppighet av VES, men sammenhengen med kardiovaskulær død og plutselig død er dårlig dokumentert. Pasienter med strukturell hjertesykdom har økt mortalitet sammenliknet med hjertefriske, og hvorvidt VES er et uttrykk for graden av patologi eller i seg selv har prognostisk verdi, er i stor grad ukjent. F.eks. har pasienter med hypertensjon og venstre ventrikkelhypertrofi ved ekkokardiografi flere VES enn hypertensive pasienter uten hypertrofi (12), men den kausale sammenhengen og prognostiske verdien er usikker. Også pasienter med hypertrofisk kardiomyopati har høy forekomst av VES, men heller ikke for disse pasientene har VES noen etablert prediktiv verdi (13).
Lown og Wolfs velkjente hierarkiske inndeling av VES stammer fra observasjoner på 60-tallet av økt forekomst av VES og plutselig død i akuttfasen etter hjerteinfarkt, men har ikke blitt validert senere. Senere studier fant også en assosiasjon mellom VES i kronisk fase etter infarkt og mortalitet (14), men dette er ikke bekreftet etter innføring av standard invasiv revaskularisering, ACE-hemmere og betablokkere. For pasienter med etablert kronisk hjertesvikt synes imidlertid forekomsten av VES å være en prediktor for kardiovaskulær død også i nyere studier, selv etter statistisk justering for ejeksjonsfraksjon og bruk av betablokker (15).
Utredning med utgangspunkt i VES må på den ene siden skille symptomfrie pasienter fra pasienter med symptomer som kan tilskrives VES, og på den andre siden skille hjertefriske pasienter fra pasienter med hjertesykdom (tabell 1). Anamneseopptaket bør søke å avdekke familiær disposisjon for iskemisk hjertesykdom, kardiomyopatier og plutselig død. Det er også viktig å spørre om epilepsi i nær familie siden kardielle synkoper kan ha blitt mistolket som epileptiske anfall. Noen ionekanalsykdommer disponerer også for både epilepsi og arytmi. Videre bør pasientens egne symptomer gjennomgås på alminnelig vis, dvs. palpitasjoner, nærsynkoper, synkoper, brystsmerter, dyspné etc.
Ofte blir VES observert i 12-avlednings-EKG eller ved korte rytmeopptak. 12-avlednings-EKG i hvile hører uansett med for alle pasienter for å identifisere tegn til iskemi, strukturell hjertesykdom, eller kanalopatier. Enkeltstudier har vist at korte observasjonsperioder (minutter) kan si noe om frekvensen av VES over en lengre periode (døgn) (16), og andre frekvensbaserte parametere enn døgnregistrering kan også ha tilleggsverdi. De fleste studier har likevel brukt opptak over 24 timer. Frekvensen av VES varierer gjennom døgnet, og 24-timers Holter-registrering gir en mulighet for å assosiere VES med aktivitet. Man bør registrere relasjonen mellom VES og egenrapporterte symptomer og sammenhengen mellom VES, aktivitet og hjertefrekvens. Dessuten bør antall VES per døgn registreres, VES-morfologien beskrives som uttrykk for ett eller flere foci og koblingsintervallet til forutgående sinusslag bemerkes. Frekvensen bør vurderes ut fra pasientens alder (7), men generelt sier den lite i seg selv med mindre frekvensen når et nivå som kan indikere risiko for VES-indusert kardiomyopati, dvs 10 000-20 000 per døgn (3).
Med opplysninger fra anamnese, 12-avlednings-EKG og 24-timers-Holter-registrering kan man korrelere evt. symptomer til VES, og gjøre en vurdering av risikoen for at det foreligger hjertesykdom eller om VES er registrert som et isolert fenomen hos en hjertefrisk pasient. Terskelen bør være lav for å supplere med ekkokardiografi for ytterligere å minske sannsynligheten for strukturell hjertesykdom, spesielt ved symptomgivende VES, høy frekvens av VES for alder eller ved mistanke om strukturell eller iskemisk hjertesykdom ut fra anamnese eller funn. I tillegg til ekkokardiografi er AKG eller annen stressbelastning ofte relevant, både for å sannsynliggjøre iskemi, men også for å avdekke evt. belastningsutløste VES eller andre arytmier.
Som beskrevet tidligere foreligger det ingen dokumentasjon for hvilken negativ prediktiv verdi normale funn ved slik utredning egentlig representerer mht. plutselig død som følge av ventrikulær arytmi utløst av VES. Den kvantitative diagnostiske verdien er derfor i realiteten knyttet til sannsynligheten for f.eks. iskemisk hjertesykdom vurdert ut fra alminnelige retningslinjer, eller hypertrofi i EKG som har 90 % positiv prediktiv verdi for hypertrofi ved ekkokardiografi. Ved funn som tyder på underliggende hjertesykdom bør videre utredning målrettes, f.eks. med MR ved mistanke om arytmogén høyre ventrikkelkardiomyopati (ARVC) eller genetisk rådgivning og evt. -testing ved familieanamnese som tyder på arvelige kardiomyopatier eller ionekanalsykdommer.
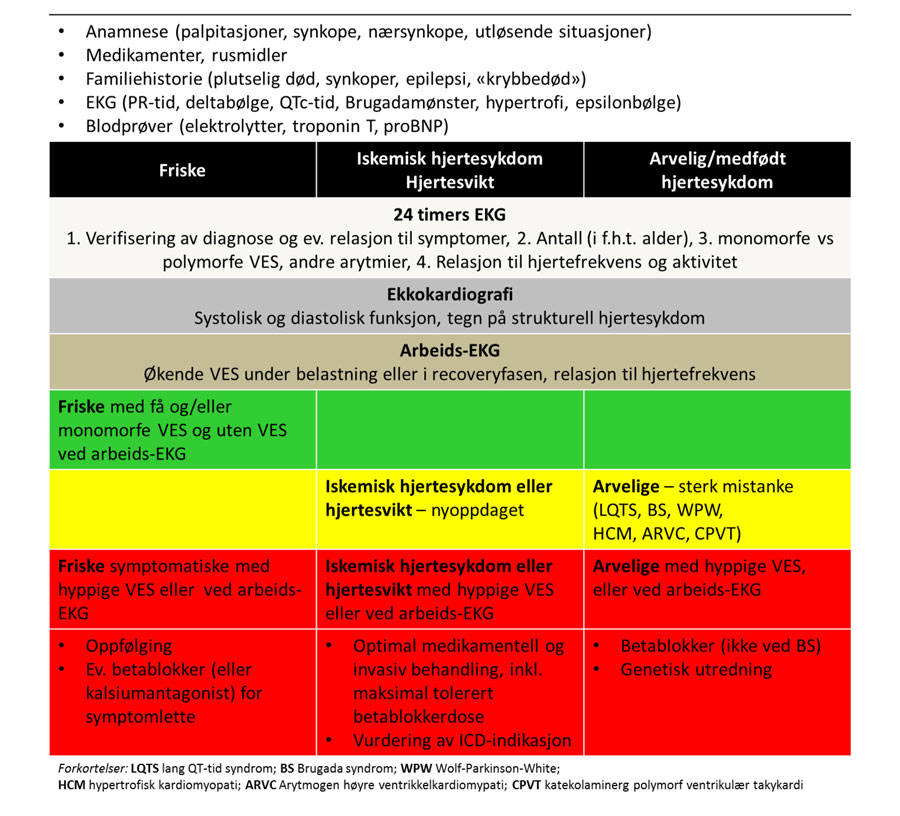
Tabell 1. Artikkelforfatterens forslag til utredning og risikostratifisering av pasienter med VES. Grønn representerer pasienter med relativt sett laveste risiko, gul intermediær risiko og rød høyeste risiko.
Behandling av VES har vært et kontroversielt tema siden CAST-studien på 1980-tallet. Studien inkluderte pasienter som hadde gjennomgått hjerteinfarkt og hadde svekket venstre ventrikkelfunksjon. I en gruppe pasienter hvor flekainid eller enkainid effektivt reduserte frekvensen av VES var mortaliteten høyere enn i en tilsvarende gruppe som ble behandlet med placebo. Studien illustrerte den proarytmiske effekten av antiarytmika og var avgjørende for hvordan senere kliniske studier har definert primære endepunkter. Siden har CAST-resultatene imidlertid også blitt brukt som argument mot en kausal sammenheng mellom VES og arytmidød, selv om studien ikke var designet til å besvare et slikt spørsmål. Videre er det verdt å merke seg at en senere subgruppeanalyse viste at pasienter hvor VES ble redusert med kombinasjon av betablokker og flekainid faktisk hadde redusert arytmidød. Det foreligger fortsatt ingen studie som har vist at å redusere frekvensen av VES forebygger arytmidød.
En konsensusrapport fra hjerterytmeforeningene i Asia, USA og Europa publisert i 2014 anbefaler kun behandling av VES for symptomlette eller hvor svært høy frekvens av VES antas å være årsak til redusert venstre ventrikkelfunksjon. For symptomlette anbefales forsøk med betablokker eller kalsiumantagonist, selv om dokumentasjon for slik behandling mangler. En studie som inkluderte pasienter med et svært høyt antall VES med venstre grenblokksmønster og inferior akse, altså forenlig med fokus i høyre utløpstraktus, viste at atenolol reduserte antall VES (17). Placebo ga imidlertid like god symptomlindring. En annen studie differensierte pasienter på bakgrunn av relasjonen mellom VES og forutgående RR-intervall (18). Nadolol reduserte frekvensen av VES hos alle pasienter med takykardiutløste VES (gjennomsnittlig 88 % reduksjon), mens en slik effekt ble observert hos færre pasienter som ikke hadde en tydelig eller blandet relasjon mellom hjertefrekvens og VES, og hos enda færre pasienter med bradykardiutløste VES. Det er likevel viktig å huske at mange pasienter med hyppige VES har annen indikasjon for farmakologisk behandling som kan redusere antall VES, f.eks. betablokker ved strukturell hjertesykdom, iskemisk hjertesykdom eller ionekanalsykdommer.
Radiofrekvensablasjon for VES brukes som et alternativ til medikamentell behandling hos utvalgte pasienter, og et stadig stigende antall publikasjoner om denne indikasjonen foreligger etter beskrivelsen av VES-indusert kardiomyopati. Den nevnte internasjonale konsensusrapporten anbefaler ablasjon til «utvalgte pasienter» som forblir svært symptomplaget over tid eller hvor et høyt antall VES forbindes med svekket venstre ventrikkelfunksjon. En meta-analyse av 15 studier hvor ablasjon ble utført for svært hyppige VES viste at ablasjon ofte hadde varig effekt med bedret venstre ventrikkelfunksjon, spesielt hos pasienter med klart redusert ejeksjonsfraksjon før ablasjonen (19). Kun tre av de inkluderte studiene var prospektive, og det foreligger ingen randomiserte studier av effekten av ablasjon. En publikasjon av retrospektivt analyserte data fra Mayoklinikken presenterer resultater av både farmakologisk behandling og ablasjon (20). Studiedesignet gjør at man skal være forsiktig med å
trekke konklusjoner mht. sammenlikning av de to
behandlingsstrategiene, men viser at en stor andel pasienter med mange VES per døgn kan hjelpes av begge strategier. Viktigere er kanskje differensieringen av pasientene på bakgrunn av grunnsykdom, kontraktil funksjon, fokus for VES og utprøving av flere forskjellige medikamenter, inkl. antiarytmika. Denne tankegangen bør åpne for mer differensierte spørsmål i fremtidige randomiserte studier og forhåpentlig mer målrettet og tilpasset behandling.
VES er et svært vanlig fenomen som kan være godartet hos reelt hjertefriske, markere underliggende hjertesykdom eller i seg selv svekke hjertefunksjonen over tid. Klinikerens oppgave er å identifisere de pasientene som trenger behandling for underliggende hjertesykdom eller fordi frekvensen av VES i seg selv kan resultere i VES-indusert kardiomyopati. Utover dette er indikasjonen for behandling av VES per i dag kun avhengig av symptomplager. Likevel vet vi at VES ofte er utløsende hendelse for maligne arytmier og plutselig død. Fremtidig forskning må identifisere pasienter hvor VES og aspekter ved VES innebærer høy risiko for slike hendelser, og hvilken forebyggende behandling som er mest effektiv for disse utvalgte pasientene.



